Achromatopsia: Understanding the rare inherited retinal disease


A recent global survey shows achromatopsia remains relatively misunderstood and underdiagnosed, and individuals with this disease face a difficult journey in pursuit of an early and accurate diagnosis.
In 1997, a book published by neurologist Oliver Sacks, MD, “The Island of the Colorblind,” revealed an unusual trait in the inhabitants of the small Micronesian island of Pingelap-they were born completely colorblind. Known on the island as maskun, which literally translates as “not see” in Pingelapese, the rare genetic disorder is better known as achromatopsia by the rest of the world.
Achromatopsia is an autosomal recessive disease that affects approximately 1:30,000 individuals and is associated with complete loss of cone function. It is most commonly caused by mutations in the CNGB3 and CNGA3 genes and is associated with severely reduced visual acuity and extreme photosensitivity, resulting in daytime blindness.
Due to a loss of cone cell function, patients have complete loss of color discrimination. Most patients with achromatopsia have an average visual acuity of 20/200, resulting in a diagnosis of legal blindness. Profound sensitivity to light during the day results in significant impairment in visual function, and many patients cope by wearing darkly tinted glasses to lessen the effect of light sensitivity.
Even though this inherited retinal disease was the focus of a popular book by the well-known Dr. Sacks, a recent global survey conducted by Achroma Corp. shows that this disease remains relatively misunderstood and underdiagnosed, and individuals with achromatopsia face a long and difficult journey in pursuit of an early and accurate diagnosis.
Diving deeper
In an effort to better understand and engage the achromatopsia community, the “Understanding the Achromatopsia Patient Experience” survey was conducted online in January 2018 on behalf of Achroma Corp. and in partnership with Applied Genetic Technologies Corp. (AGTC), a gene therapy company. The survey, distributed through Achroma Corp.’s network, was completed by 226 respondents who have been diagnosed with-or have a child who has been diagnosed with-achromatopsia.
Initial symptoms of achromatopsia typically appear in infancy, as this is a congenital disorder. Symptoms can include nystagmus (rapid involuntary eye movements), as well as photosensitivity and markedly reduced visual acuity.
According to the global survey, photosensitivity is reported as being the most debilitating and bothersome symptom of achromatopsia; on a 0–100 scale, adults with achromatopsia give photosensitivity a rating of 77 on severity and 75 on being bothersome.
This severe photoaversion significantly and negatively impacts their ability to function daily and takes an emotional toll on their health and wellness.
The majority of affected individuals reported that their photosensitivity had not changed over time (53% of adults and 82% of children).
However, more than one-third of adults believed that their photosensitivity had worsened over time. These individuals reported that they had taken additional steps to adapt and function. Primarily, 58% of individuals adapted by using eyewear with a darker tint or extreme gradient, 53% expanded their use of eyewear and 44% avoided the outdoors
Regarding diagnosis, most patients in the survey described a long and circuitous route to correct diagnosis, with a relatively longer course to diagnosis for adults as compared to the children. Parents typically pursue answers from their general or pediatric healthcare providers, and see an average of four healthcare providers in a span of three years before receiving a diagnosis for their child, with 68% taking more than a year to receive a diagnosis after the initial onset of symptoms
Twenty-three percent of children still received an incorrect diagnosis of retinal or cone dystrophy before being accurately diagnosed with achromatopsia. Adults with achromatopsia usually see an average of seven healthcare providers over a span of more than five years to receive the correct diagnosis. More than one third of these individuals were misdiagnosed with retinal or cone dystrophy before receiving the correct diagnosis of achromatopsia.
The survey results indicate that only 58% of adults and 65% of children with achromatopsia have received genetic testing to confirm the correct diagnosis and the underlying gene responsible.
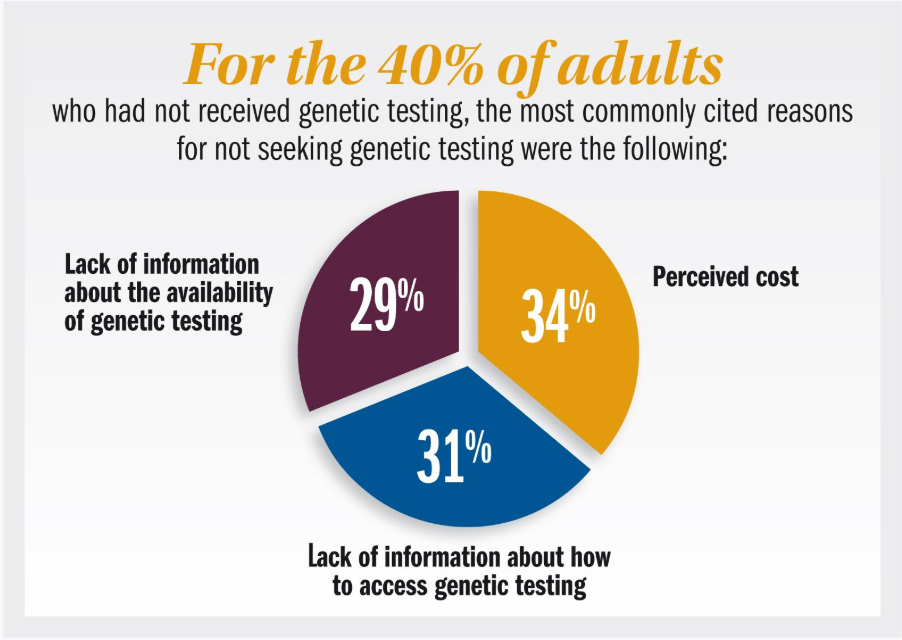
In the past, the lack of genetic confirmation of disease could be blamed on the lack of therapeutic options. Due to this, many ophthalmologists and their achromatopsia patients resigned themselves to simply managing the disease and never sought out genetic testing to secure or confirm a correct diagnosis. For the 40% of adults who had not received genetic testing, the most commonly cited reasons for not seeking genetic testing were the following:
- Perceived cost (34%)
- Lack of information about how to access genetic testing (31%)
- Lack of information about the availability of genetic testing (29%)
For parents of children with achromatopsia who had not received genetic testing, the most commonly cited reason was lack of information about accessing genetic testing (27%).
However, even in the past few years, the landscape for achromatopsia has radically changed. With several gene therapy studies in achromatopsia under way, it becomes imperative that ophthalmologists have a conversation with their inherited retinal disease patients about receiving genetic testing.
Currently, members of the Foundation Fighting Blindness (FFB) registry who reside in the United States and have a clinical diagnosis of an orphan inherited retinal dystrophy studied by the foundation can participate in a free genetic testing and ocular genetic counseling study with the assistance of their eye care professional. This research study, which is IRB-approved and available for a finite time, is available through the FFB registry (www. myretinatracker.org).
By knowing their specific gene mutation, achromatopsia patients, as well as others living with inherited retinal diseases, may have the opportunity to participate in applicable clinical trials. These clinical trials are investigating potential treatments for the condition while also advancing the scientific understanding of achromatopsia.
AGTC is currently recruiting for two phase I/II clinical trials for individuals with achromatopsia caused by mutations in either the CNGB3 or the CNGA3 gene.
Information about the phase I/II clinical trial in achromatopsia caused by CNGA3 can be found at ClinicalTrials.gov under the trial identifier number NCT02935517, while the phase I/II clinical trial in achromatopsia caused by CNGB3 can be found under the trial identifier number NCT02599922.
Author’s Note: Regarding these survey results, it is important to note that the survey was completed by those who were fortunate enough to receive a correct diagnosis and therefore become involved in Achroma Corp.’s network (www.achromacorp.org). Due to selection bias, the rates of misdiagnosis and barriers to genetic testing are likely much higher than captured by this survey.
Disclosures:
Christine N. Kay, MD
P: 352/371-2800
Dr. Kay is a principal investigator in the AGTC-sponsored gene therapy trials for CNGA3 and CNGB3 achromatopsia.
References:
1. Sharpe LT, Stockman A, Jagle H, Nathans J. Opsin genes, cone photopigments, color vision, and color blindness. In: Gegenfurtner K, Sharpe LT, eds. Color Vision: from Genes to Perception. Cambridge, UK: Cambridge University. Press; 1999:3-52.
2. Achroma Corp. announces global survey results of 226 people with achromatopsia. Achroma Corp. https://www.achromacorp.org/PatientJourney.html. Accessed June 27, 2018.
Newsletter
Keep your retina practice on the forefront—subscribe for expert analysis and emerging trends in retinal disease management.











